Key Points
- Sickle cell disease (SCD) refers to a group of hemoglobinopathies characterized by the formation of long chains of hemoglobin when deoxygenated within capillary beds. This results in sickle-shaped red blood cells (RBCs) and multiorgan dysfunction.
- The complications from SCD result from two overlapping subtypes: vaso-occlusion and chronic hemolysis from endothelial dysfunction.
- SCD is characterized by repeated episodes of severe painful crisis and acute chest syndrome. Other complications include stroke, priapism, chronic pain, avascular necrosis, pulmonary hypertension, and leg ulcers.
- This is part I of a two-part series that will cover the SCD variants, pathophysiology, and systemic effects. Part II will review key aspects of the perioperative management of patients with SCD.
Introduction
- Hemoglobin is a protein in RBCs that transports oxygen from the lungs to all the organs and tissues in the body.
- There are three types of normal hemoglobin:
- Hemoglobin A (HbA) is the most common form of adult hemoglobin, composed of two alpha subunits and two beta subunits (α2β2).1 After about six weeks of life, HbA represents about 95% of hemoglobin in normal individuals.
- Hemoglobin A2 (HbA2) consists of two alpha and two delta chains (α2ƍ2). HbA2 is a minor form of hemoglobin that accounts for about 2% of the hemoglobin of normal adults.1
- Hemoglobin F (HbF) consists of two alpha and two gamma chains (α2γ2). HbF is the dominant hemoglobin until about six weeks of age.
SCD
- SCD is an umbrella term for a group of hemoglobinopathies in which abnormal hemoglobin results in sickled RBCs and potential multisystem dysfunction.1 They are inherited in an autosomal recessive pattern (Figure 1 and Table 1).
- SCD is caused by a single nucleotide mutation of the β-globin gene.
- Hemoglobin S (HbS) is formed by glutamate being substituted by valine.
- Hemoglobin C (HbC) is formed by glutamate being substituted by lysine.
- SCD is caused by the coinheritance of HbS and other variant hemoglobins that result in the sickling of RBCs, chronic hemolytic anemia, and vaso-occlusive crises.1
- Sickle cell anemia results from inheriting a HbS gene from each parent. These individuals are homozygous HbSS and have the most severe form.
- Hemoglobin SC disease (HbSC) is caused by the co-inheritance of HbS and HbC. HbSC accounts for nearly 30% of SCD in the United States and the United Kingdom.2
- HbC disease (HbCC) is caused by a dual inheritance of HbC. These patients do not have sickling but do have chronic hemolytic anemia.1
Mild Variants
- Sickle cell trait (HbAS) results from inheriting one normal hemoglobin (HbA) and one mutated HbS. These individuals are heterozygous carriers (HbAS) and usually do not have any symptoms. However, they have an increased risk of chronic kidney disease, decreased renal concentrating ability, hematuria, venous thromboembolism, splenic infarction, and sudden death on exertion. They are also at an increased risk of sickling if hypoxia (PaO2 < 45mmHg is the threshold for sickling), hypovolemia, or acidosis occurs.
- Hemoglobin C trait results from inheriting hemoglobin C in addition to one normal HbA. These patients are phenotypically normal.
- HbS/β-thalassemia is caused by co-inheritance of HbS and beta-thalassemia mutations. It is also considered a mild variant, but symptoms may vary based on the genetic makeup.

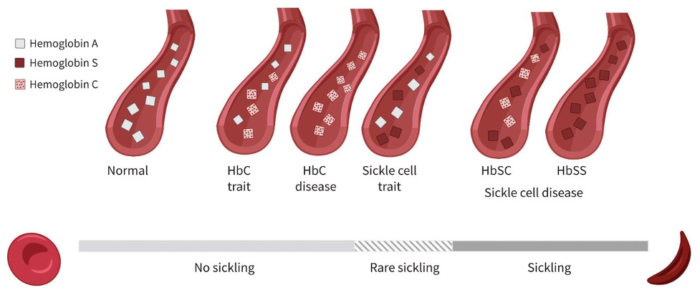
Figure 1. Hemoglobin variants and the degree of sickling
The degree of red blood cell sickling depends on the presence of at least one normal hemoglobin allele. Individuals with sickle cell trait (HbS) or HbC trait (HbC) have a lower risk of sickling than those with two variant alleles, such as HbSS or HbSC. Used with permission from McFarland T et al. Unrecognized hemoglobin SC sickle cell disease complicated by sepsis and cholestasis. CMAJ. 2022;194(17):608-11.
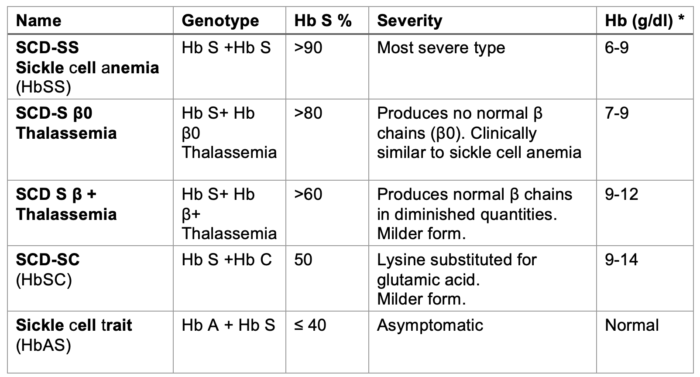
Table 1. SCD types. *Hb levels in the absence of blood transfusion in the past four months indicate the clinical severity of the disease.
Epidemiology and Prognosis
- SCD affects millions of people worldwide but is predominantly found in those with ancestry from sub-Saharan Africa, the Middle East, and India. Worldwide, approximately 300,000 babies are born with severe hemoglobin disorders each year.3
- In the United States, SCD affects nearly 100,000 people. Approximately 1 in 13 African Americans have HbAS, and 1 in 365 African Americans have SCD.4
- Until the 1990s, up to 30% of children with SCD died from infections. Early diagnosis, vaccinations, antibiotics, and education have reduced this number to below 3%.4
- The current estimated life expectancy for patients with SCD in the United States is 54 years. In contrast, 50-90% of children with SCD die before age five years in sub-Saharan Africa.5
Pathophysiology and Complications
Pathophysiology
- The pathology of SCD begins with HbS polymerization. This polymerization of hemoglobin alters the shape and physicochemical features of RBCs.
- Sickled RBCs have increased adhesion to endothelial walls, leading to endothelial damage and rapid hemolysis. This interaction between the endothelium, white blood cells, and platelets leads to an endovascular inflammatory cascade.5
Complications
- The complications of SCD can be divided into two overlapping subtypes with distinct mechanisms- increased vaso-occlusion and endothelial dysfunction from decreased nitric oxide bioactivity.6
- Vaso-occlusion or obstruction of blood flow occurs due to microvascular entrapment of sickled RBCs and leukocytes. Vaso-occlusion leads to complications such as acute pain crisis, acute chest syndrome, and avascular necrosis.5,6
- In the second subtype, decreased nitric oxide bioactivity and endothelial dysfunction cause chronic intravascular hemolysis, which can eventually lead to stroke, pulmonary hypertension, priapism, etc.5,6
- Symptoms of SCD typically begin early in life, around five months. The most common symptoms include anemia, pain crises, acute chest syndrome, bacterial infections, swelling in the hands and feet, recurrent priapism, and avascular necrosis.
- Over time, patients often develop chronic conditions such as chronic kidney disease, pulmonary hypertension, chronic pain, deep vein thrombosis, and vision loss.
- Overall, the clinical severity of sickle cell anemia varies among individuals. Factors that may determine the severity of complications include fetal hemoglobin expression and the co-inheritance of other hemoglobin variants, such as alpha thalassemia.5
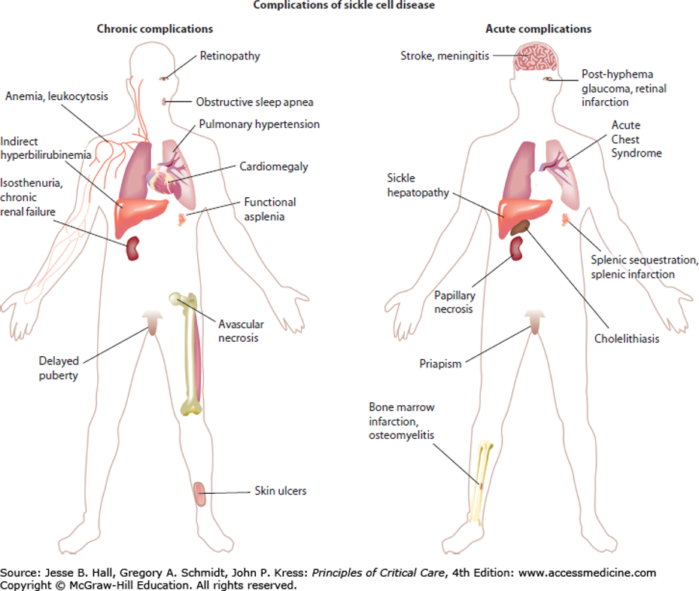
Figure 2. Major complications of SCD. Used with permission from Kato GJ, Gladwin MT. Sickle cell disease. In: Hall JB, Schmidt GA, Kress JP. eds. Principles of Critical Care, 4e. McGraw Hill; 2014. Figure 96-1.
Vaso-occlusive Crisis
- A vaso-occlusive crisis is the culprit of many of the complications related to SCD and can occur in various organs.
- Vaso-occlusion of the pulmonary vasculature is the cause of acute chest syndrome, which is one of the leading causes of morbidity and hospitalizations in patients with SCD.6
- When vaso-occlusion occurs in the bone, it can lead to bone tissue death called avascular necrosis. This most commonly occurs in the hip joint but can also occur in other areas such as the spine, shoulders, and ankles.
- Trapping of sickled RBCs can lead to acute and painful enlargement of the spleen. This is referred to as a splenic sequestration crisis. Most patients will have spleen infarctions before the end of childhood.
Acute Pain Crisis
- As one of the cardinal features of SCD, acute pain crisis is a presenting symptom for many sickle cell patients. The obstruction of blood flow and tissue ischemia lead to acute and severe pain. Neuropathic and central pain pathways are also involved in the complex pathophysiology of pain in SCD.5
- Pain can occur in any part of the body but frequently affects the extremities, back, and chest. The intensity of pain varies and can last a few hours to several days. The frequency of pain crises is highly variable. Some people have only a few pain crises a year, while others can have several a year.
- Management of acute pain crisis involves opioids, nonsteroidal anti-inflammatory drugs (NSAIDs), and rehydration.5 Low-dose ketamine infusions and regional anesthesia may be considered for opioid-refractory pain. Supplemental oxygen and RBC transfusions are not recommended for the routine management of painful crises.5
- Hydroxyurea is a myelosuppressive drug used as a preventive treatment for SCD. It has been shown to reduce the frequency of painful episodes and increase the hemoglobin level.8
Acute Chest Syndrome
- Acute chest syndrome is the second most common cause of hospitalization among patients with SCD.6 Three major causes of acute chest syndrome have been proposed.6
- Pulmonary infection with a community-acquired pathogen, which incites an excessive inflammatory response
- Embolization of bone marrow fat
- Intravascular pulmonary sequestration of sickled RBCs leads to further ischemia and endothelial damage
- All three triggers cause lung injury and infarction, resulting in ventilation-perfusion mismatch and hypoxemia, which further exacerbates the polymerization of sickled RBCs.6
- The presenting symptoms include fever, cough, hypoxia, tachypnea, chest pain, and wheezing. The diagnosis can be difficult due to its clinical similarities to pneumonia.
- The diagnosis of acute chest syndrome requires a new infiltrate on chest imaging plus any two of the following: pleuritic chest pain, hypoxemia, tachypnea, or fever.5-7
- Risk factors for acute chest syndrome include young age, presence of asthma, smoking, recent trauma or surgery, and low concentration of fetal hemoglobin.7
- Management includes broad-spectrum antibiotics, exchange transfusion, and supportive care such as supplemental oxygen, pain management, and hydration.
- The mean length of hospitalization for adults with acute chest syndrome is 10.5 days, compared with 3-4 days for uncomplicated vaso-occlusive pain crises.6
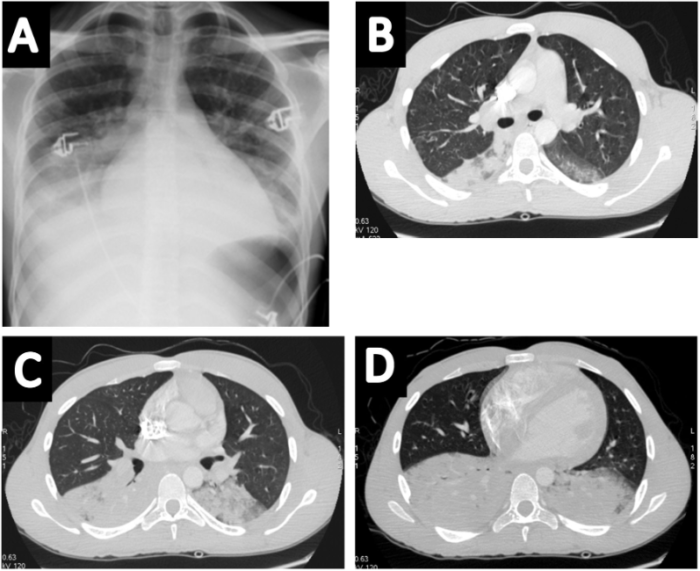
Figure 3. Images of a patient with acute chest syndrome and bilateral consolidations predominating at the bases on chest radiograph (A) and CT (B-D)
Used with permission from Mekontso DA et al. Lung imaging during acute chest syndrome in sickle cell disease: Computed tomography patterns and diagnostic accuracy of bedside chest radiograph. Thorax 2014; 69:144-151.
Stroke
- The sickling of RBCs significantly increases the risk of stroke. SCD is the leading cause of ischemic stroke in children.5 By age 20, approximately 11% of patients have clinical evidence of a stroke.9 By age 45, 24% of patients have suffered from a stroke.9
- Patients are at risk for hemorrhagic strokes, acute ischemic strokes, chronic ischemic strokes, and silent infarctions.9 The recurrence rate of stroke in SCD is 66%.9
- Transcranial Doppler is a noninvasive method of measuring blood flow in the intracranial arteries. Elevated velocity in the intracranial arteries is a strong predictor of stroke risk. Therefore, transcranial Doppler can be used as a screening test to help identify patients who are at high risk for stroke.9
- Measures to prevent stroke include routine blood transfusions along with overlapping hydroxyurea therapy and intermittent phlebotomy to remove iron.9
Acute Anemia
- Acute anemia in SCD is defined as an acute decrease in hemoglobin from baseline by 2 g/dL or greater. Splenic sequestration of sickled RBCs in the spleen is the most common cause of anemia and presents as left upper quadrant pain with splenomegaly, decreasing hemoglobin, and increased reticulocyte counts. Treatment includes RBC transfusion to maintain euvolemia and oxygenation.5
- Aplastic crisis is a second cause of anemia, characterized by an acute decrease in Hb and a significant decrease in reticulocyte count from baseline. It is commonly caused by a parvovirus B19 infection or an acute inflammatory illness.5
Priapism
- Priapism, or a painful erection lasting more than 4 hours, affects approximately 40% of males with SCD. Low blood flow due to venous congestion impedes blood outflow and causes priapism. Treatment includes supportive measures (oral fluids, warm showers, walking) followed by intravenous fluid, analgesia, and local injection of sympathomimetics if needed.5
Venous Thromboembolism
- Venous thromboembolism is common in SCD, and pharmacological prophylaxis is indicated for hospitalized adults and at-risk children with SCD.
Pulmonary Hypertension
- Pulmonary hypertension develops secondary to the progressive obstruction of small pulmonary arterioles and endothelium changes due to oxidative stress.10 This culminates with increased pulmonary vascular resistance and pulmonary arterial hypertension.
- The severity of anemia and rate of hemolysis are both risk factors for the development of pulmonary hypertension.10
- Reduced exercise capacity is one of the key clinical findings. Diagnosis can be made based on history, assessment of exercise tolerance tests, and Doppler echocardiography revealing elevated tricuspid valve regurgitation jet. A definitive diagnosis of pulmonary hypertension is determined with a right heart catheterization finding of elevated pulmonary pressures.10
- Pulmonary vasodilators are the mainstay of treatment for pulmonary arterial hypertension caused by SCD. Sildenafil, a phosphodiesterase inhibitor, is a commonly prescribed drug that promotes pulmonary vasodilation. Other drugs include prostacyclin agonists (epoprostenol) and endothelin receptor antagonists (bosentan).10
Renal Manifestations
- Renal disease in SCD is characterized by hyperperfusion in the renal cortex, hypoperfusion in the renal medulla, and an exaggerated stress-induced vasoconstrictive response and accounts for 16-18% of mortality in SCD.11
- Glomerular disease usually manifests as focal segmental glomerulosclerosis in SCD.
- Acute kidney injury (AKI) develops in 4-10% of hospitalized patients and is more common in patients with acute chest syndrome than in those with painful crises. Hypovolemia, infections, NSAIDs, and rhabdomyolysis can increase the risk of AKI.
- Urine concentrating ability may be impaired in very young children, including infants with SCD, predisposing them to dehydration. Most adults with SCD are unable to achieve a urine osmolarity above 450mOsm/kg.
- Normokalemic renal tubular acidosis, as well as hyperkalemic, hyperchloremic metabolic acidosis, may occur in SCD.
- Proteinuria and hematuria are also common. Proteinuria becomes more frequent with age, with 27% of SCD patients developing proteinuria in the first three decades of life. Patients with low levels of hemoglobin, hypertension, a history of acute chest syndrome, prior vaso-occlusive crisis, pulmonary hypertension, stroke, and parvovirus infection are more likely to have proteinuria. Hematuria occurs because of capillary congestion with extravasation of RBCs into the tubular lumen, papillary necrosis, and, in rare instances, renal medullary carcinoma. Hematuria can be microscopic or macroscopic. Macroscopic hematuria can be life-threatening.11
SCD Management
- The treatment goals of SCD are symptom control and management of disease complications. Treatment primarily focuses on supportive care, such as providing oxygen and antibiotics, as indicated, transfusing blood, and managing pain.5
- Opioids are the mainstay of pain management in SCD. They effectively treat acute and chronic pain. Careful monitoring is required to avoid significant side effects such as respiratory depression, nausea, vomiting, constipation, tolerance, and dependence.
- NSAIDs are commonly used to treat pain in SCD. They help to reduce the inflammation in vaso-occlusive bone pain. These medications must be used judiciously, given the associated risk of renal, gastrointestinal, and cardiovascular toxicity.
- Chronic blood transfusions serve as both a therapeutic and preventive method for managing complications related to SCD. They are also the standard of care for managing hemolytic crises, acute chest syndrome, acute pain crises, and stroke.
- Hydroxyurea is commonly used as first-line therapy for preventing complications such as acute chest syndrome in patients with a history of vaso-occlusive events. It works by inducing fetal hemoglobin (HbF) production, which inhibits HbS polymerization, reducing red cell rigidity and hemolysis. In addition, hydroxyurea increases nitric oxide, decreases red cell adhesion, and decreases leukocytes.5 It has proven efficacy in reducing vaso-occlusive crises, reducing hospitalizations, and improving overall survival.8
- In addition to hydroxyurea, three new drugs have been approved in the United States for SCD.5
- L-glutamine is an oral amino acid supplement that decreases reactive oxygen species in RBCs, thereby reducing sickling and adhesivity.
- Crizanlizumab is a monoclonal antibody directed against P-selectin, an adhesion molecule found on activated platelets and endothelial cells that mediates vaso-occlusion.
- Voxeltor stabilizes the oxygenated Hb in SCD by promoting HbS binding to oxygen, thereby decreasing HbS polymerization and related hemolysis.
- Hematopoietic stem cell transplantation has demonstrated efficacy as a curative treatment for SCD patients.8 However, it requires strict human leukocyte antigen (HLA) matching, which can make finding a donor difficult. Also, there is a potential for graft rejection, graft versus host disease, and disease relapse. These transplants are rare, given the issues with finding suitable donors and the aforementioned associated risks.
- Gene therapy is another promising advancement that could lead to a cure.8 In December 2023, the FDA approved two new cell-based gene therapies for SCD with recurrent vaso-occlusive crises – Casgevy and Lyfgenia for patients 12 years and older.12
- Casgevy uses CRISPR technology to edit the genome to produce HbF at higher levels. Approval was based on 29/31 eligible patients achieving freedom from the severe vaso-occlusive crisis for at least 12 consecutive months during 24 months of follow-up. Lyfgenia uses a lentivirus to genetically modify cells to produce more HbAT87q, a type of adult hemoglobin. Approval was granted based on the complete resolution of vaso-occlusive crises between 6 and 18 months after treatment in 28/32 patients.
- Side effects of these drugs include low platelet and white blood cell counts, nausea, abdominal pain, vomiting, stomatitis, and febrile neutropenia. Lyfgenia has a black box warning regarding the risk of developing a hematological malignancy. Both medications received an orphan drug, fast track, regenerative medicine advanced therapy, and priority review.
References
- Sheehan VA, Gordeuk VR, Kutlar A. Disorders of hemoglobin structure: Sickle cell anemia and related abnormalities. In: Kaushansky K, Prchal JT, Burns LJ, Lichtman MA, Levi M, Linch DC. eds. Williams Hematology, 10th edition. McGraw Hill; 2021.
- Pecker L, Schaefer BA, Luchtman-Jones L. Knowledge insufficient: The management of haemoglobin SC disease. Br J Haematol. 2017. 176 (4): 515-26 PubMed
- World Health Organization. Sickle Cell Disease. Accessed August 2, 2024. Link
- Center for Disease Control. Data and Statistics on Sickle Cell Disease. Accessed August 2, 2024. Link
- Kavanagh PL, Fasipe TA, Wun T. Sickle cell disease: A review. JAMA. 2022. 328(1):57-68. PubMed
- Gladwin MT, Vichinsky E. Pulmonary complications of sickle cell disease. N Engl J Med. 2008; 359: 2254-65. PubMed
- Paul RN, Castro OL, Aggarwal A, et al. Acute chest syndrome: sickle cell disease. Eur J Haematol. 2011 Sep;87(3):191-207. PubMed
- Tisdale JF, Thein SL, Eaton WA. Treating sickle cell anemia. Science. 2020.13; 367(6483): 1198-1199. PubMed
- Verduzco LA, Nathan DG. Sickle disease and stroke. Blood.2009;114(25):5117-25. PubMed
- Potoka KP and Gladwin MT. Vasculopathy and pulmonary hypertension in sickle cell disease. Am J Physiol Lung Cell Mol Physiol. 2015. 308(4): L314-24. PubMed
- Nath KA, Hebbel RP. Sickle cell disease: renal manifestations and mechanisms. Nat Rev Nephrol. 2015;11(3):161-171. PubMed
- United States Food and Drug Administration. FDA News Release. FDA Approves First Gene Therapies to Treat Patients with Sickle Cell Disease. December 08, 2023. Accessed August 2, 2024. Link
Copyright Information

This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.