Key Points
- The most common presentation of esophageal atresia (EA) and tracheoesophageal fistula (TEF) is a proximal blind esophageal pouch and a distal TEF.
- EA and TEF are commonly associated with other congenital anomalies, and therefore, a comprehensive preoperative evaluation is warranted.
- Maintaining adequate oxygenation and ventilation during surgical repair can be challenging, and typically, the tip of the endotracheal tube (ETT) is positioned between the TEF and the carina.
Introduction
- EA includes a group of congenital anomalies characterized by an interruption of the continuity of the esophagus, with or without a TEF, which is an abnormal communication between the esophagus and the trachea.1 While EA and TEF may present as isolated anomalies, they often present together with an incidence ranging from 1:2500 to 1:4000 births.1-3
- Several classification systems, such as the Vogt and Gross classifications, have been used to describe the anatomical variations (Figure 1). The most common presentation in 80-85% of patients is EA with a blind esophageal pouch with a distal TEF (Gross type C, Vogt 3B).

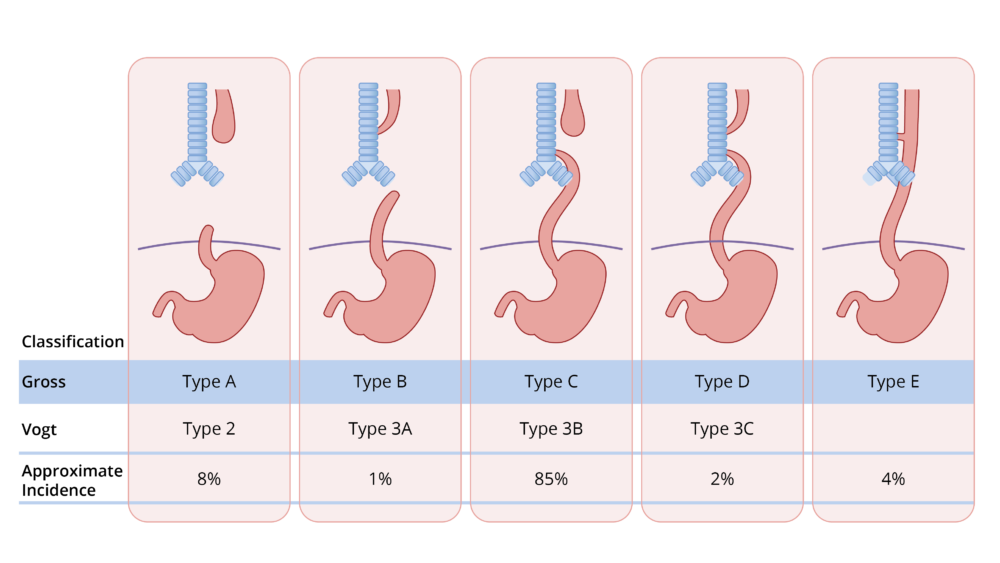
Figure 1. Gross and Vogt classification of EA and TEF.
Associated Anomalies
- About 20-30% of these patients are born prematurely, and 50% of the patients have additional congenital defects:1
- congenital heart disease: 30% (ventricular septal defect, atrial septal defect, tetralogy of Fallot, atrioventricular canal, and coarctation of the aorta)1;
- genitourinary disorders: 14% (gastrointestinal disorders), 13% (duodenal atresia, imperforate anus, malrotation), skeletal anomalies (10%) and respiratory (6%).1
- VACTERL association refers to a group of anomalies that commonly coexist in patients with EA and TEF. At least three of the VACTERL lesions are seen in 20-30% of patients with EA and TEF.1,2 The VACTERL lesions include:
- Vertebral anomalies: hemi vertebrae, scoliosis, vertebral dysgenesis.
- Anorectal anomalies: imperforate anus.
- Cardiac anomalies: VSD, ASD, TOF, AV canal, coarctation.
- Tracheo-Esophageal fistula
- Renal anomalies: renal agenesis, hydronephrosis, horseshoe kidney, urethral atresia, hypospadias.
- Limb anomalies: radial aplasia, syndactyly, polydactyly, proximally placed thumbs.
- CHARGE syndrome is another group of anomalies that may be present in patients with EA and TEF. CHARGE is an acronym for coloboma, heart defect, choanal atresia, mental and growth retardation, genital anomalies, and ear anomalies.
- Patients at high risk are premature infants with a birth weight less than 2 kg, associated cardiac anomalies, poor pulmonary compliance, and large pericardial fisutals.3
Clinical Presentation/Diagnosis
- Prenatally, EA and TEF is suspected based on the findings of maternal polyhydramnios and the absence of a stomach bubble on ultrasound.1-3 However, maternal polyhydramnios is a nonspecific sign, and the overall prenatal detection rate is about 40-50%.1,2
- Postnatally, EA and TEFs are diagnosed with the classic signs of excessive secretions, choking during feeding, and the inability to pass an orogastric tube beyond 9-10 cm. A chest x-ray shows the orogastric tube coiled in the blind esophageal pouch. Air in the stomach indicates the presence of a distal TEF. While a contrast esophagogram (Figure 1) can confirm the diagnosis of EA, it is not routinely performed.2
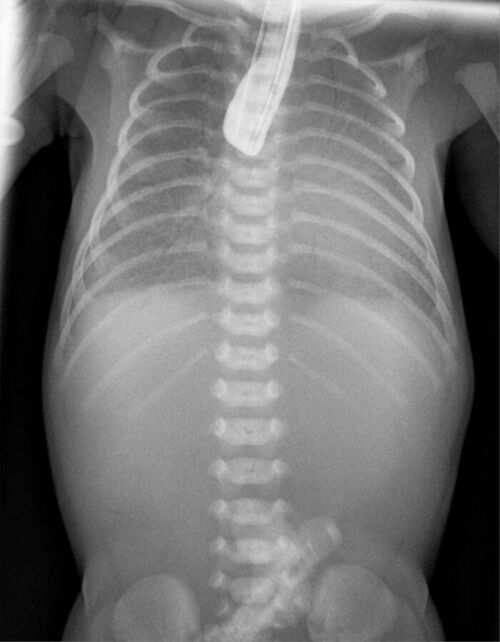
Figure 2. Contrast esophagogram in a newborn with EA showing contrast in a blind upper esophageal pouch. Source: Wikimedia Commons. Hellerhoff. CC-BY-SA-3.0.
Preoperative Management
- Prior to surgery, the patients are maintained in a 30° head-up position with a suction tube draining the esophageal pouch to minimize the possibility of aspiration of gastric contents and saliva.1,2 The surgical repair is usually considered urgent, not emergent, and is performed in the first or second day of life.
- Preoperative assessment includes a thorough evaluation of the pulmonary and hemodynamic status and a search for associated anomalies.1,2 Common preoperative investigations include:
- echocardiography to rule out cardiac defects and right-sided aortic arch (2.5% of cases)1,2;
- blood tests should include blood cell counts, electrolytes, and crossmatch1;
- chest radiographs show the typical upper mediastinal position of the nasogastric tube; air in the stomach confirms the presence of a TEF.
- ultrasound examination of the head to evaluate the presence of intracranial hemorrhage;
- ultrasound examination of the kidneys to rule out renal anomalies;
- ultrasound of the spinal cord to rule out vertebral anomalies.1
Surgical Considerations
- Traditionally, an open surgical repair is performed via a thoracotomy on the side opposite the aortic arch (usually right thoracotomy) using a posterolateral, extrapleural approach.1-3 The TEF is first ligated, followed by a primary anastomosis of the proximal and distal esophageal segments. The anesthesiologist may be asked to assist in the identification of the blind upper esophageal segment by advancing the nasogastric tube. The primary anastomosis of the esophagus is completed over a nasogastric tube that has been advanced into the distal esophageal segment or stomach. The nasogastric tube is then firmly secured to prevent accidental removal.
- Thoracoscopic repair of EA and TEF is being performed in carefully selected patients.2,3 Patients are positioned in a modified 45° prone position to allow the lung to fall away from the posterior mediastinum, allowing better surgical exposure. Carbon dioxide is insufflated at low pressures to affect lung collapse and one-lung ventilation is usually not needed.2,3 The main advantage of thoracoscopic repair is the avoidance of musculoskeletal sequelae (winged scapula, rib deformities, scoliosis, etc.) later in life and superior surgical visualization.
- If the two esophageal ends are too far apart (more than two to four vertebral bodies), a staged repair is planned.1,2 The TEF is ligated, and a gastrostomy tube is inserted for nutrition. Attempts are made to lengthen the esophagus and shorten the gap between the two esophageal segments, followed by a definitive repair at 3-6 months of age.
- Emergency ligation of the distal TEF may be necessary in preterm infants with EA/TEF and severe respiratory distress syndrome requiring mechanical ventilation.1,2 Emergency gastrostomy was traditionally performed but no longer recommended due to worsening of gas flow to the lungs. Following emergency transpleural ligation of the TEF at the bedside, definitive surgical repair can be performed in 8-10 days.1,2
Anesthetic Considerations
Monitoring
- In addition to standard ASA monitors, measurement of pre and postductal oxygen saturations and continuous hemodynamic monitoring is encouraged. If an umbilical arterial catheter is not present, a femoral or radial arterial catheter may be inserted.2
- Even though blood loss and fluid shifts are typically minimal, adequate venous access is essential, and an umbilical venous catheter is a good alternative.
- As in all neonatal cases, temperature monitoring and frequent measurement of blood gases, electrolytes, and glucose is imperative.
Bronchoscopy
- Rigid or flexible bronchoscopy is often performed prior to surgical repair to evaluate the presence, location, and size of the TEF and rule out additional fistulas.1,2
- Either an intravenous or inhalational induction can be used, and spontaneous ventilation is maintained. Typically, general anesthesia is maintained with a propofol infusion.
- Rigid bronchoscopy offers the advantage of ventilating through the scope, if necessary. Flexible bronchoscopy allows accurate positioning of the ETT distal to the TEF and proximal to the carina. It also can be used to guide the placement of a balloon Fogarty catheter to occlude the TEF.
ETT Positioning
- The tip of the ETT is positioned between the fistula and the carina using an auscultation method or fiberoptic bronchoscopy.2,4,5 Repositioning of the patient for thoracotomy and intraoperative surgical manipulation can result in ETT displacement.
- Particular attention must be paid to avoid direct intubation of the TEF. If the fistula is located just above or at the carina, options for ETT positioning include:2,4,5
- intubation of the main stem bronchus and one-lung ventilation;
- occlusion of the TEF with a Fogarty catheter;
- ETT positioned proximal to TEF, and gentle ventilation used to avoid gastric insufflation.
Ventilation
- Maintaining adequate oxygenation and ventilation during the surgical repair can be challenging, especially in neonates with preexisting respiratory compromise secondary to aspiration pneumonitis. Lateral positioning and surgical retraction of the lungs further compromise oxygenation and ventilation.1
- Avoiding excessive positive pressure ventilation before the ligation of the fistula is crucial.2 The negative intrathoracic pressure generated during spontaneous ventilation allows inspired gases to preferentially enter the lungs as opposed to flowing across the TEF. However, the decreased pulmonary reserve in these neonates often necessitates gently assisting the neonate’s ventilation. Some clinicians routinely paralyze stable patients after demonstrating adequate bag-mask ventilation and absence of gastric insufflation.4
- Hypoventilation during spontaneous ventilation frequently results in hypercarbia and can be problematic, especially in patients with associated congenital heart disease and preexisting pulmonary hypertension. Inspired oxygen concentrations might need to be temporarily increased to compensate for desaturation resulting from surgical retraction and compression of the lungs. Other causes of ventilatory issues include:
- accidental displacement of the ETT into the main stem bronchus or fistula;
- obstruction or kinking of the ETT.
- After the fistula is ligated, conventional positive pressure ventilation can be initiated.
Postoperative Management
- The decision regarding extubation in the operating room depends on several factors including prematurity, associated comorbidities, preoperative pulmonary status, ease of surgical repair, and issues with intraoperative ventilation.1 Most neonates remain intubated for 24-48 hours postoperatively.1-3 Patients who have undergone long-gap EA repairs often have tension at the anastomotic site and remain intubated and paralyzed for longer periods (5-7 days). 1,2
- Postoperative analgesia is achieved using multimodal analgesia and opioids or an epidural catheter, especially if early extubation is planned. Alternatives to epidural analgesia include local anesthetic infiltration, intercostal blocks, paravertebral catheters or intrapleural infusion of local anesthetics.2
References
- Spitz L. Oesophageal atresia. Orphanet J Rare Dis. 2007; 2:24. PubMed
- Knottenbelt G, Skinner A, Seefelder C. Tracheo-oesophageal fistula and oesophageal atresia. Best Pract Res Clin Anaesthesiol 2010; 24: 387-401. PubMed
- Holcomb GW, et al. Thoracoscopic repair of esophageal atresia and tracheoesophageal fistula. A multi-institutional analysis. Ann Surg. 2005; 242:1-9. PubMed
- Broemling N, Campbell F. Anesthetic management of congenital tracheoesophageal fistula. Paediatr Anaesth 2011; 21: 1092-99. PubMed
- Ho AM, Dion JM, Wong JCP. Airway & ventilatory management options in congenital TE fistula repair. J Cardiothorac Vasc Anesth 2016; 30: 515-20. PubMed
Other References
- Chatterjee D. Tracheoesophageal fistula. OpenAnesthesia. Published January 2017. Accessed January 25, 2023. Link
Copyright Information

This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.